On the Performances of Density Functionals for Open Shell First-Row Transition Metal Compounds
Abstract
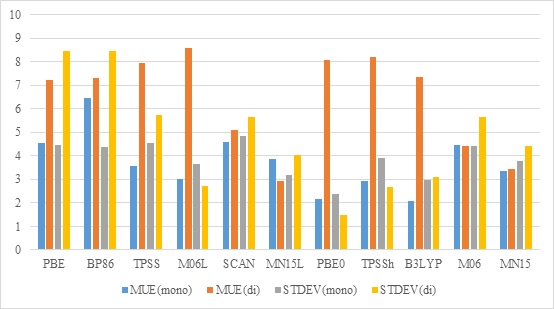
The selection of density functional is the key to obtain useful results in a computational work. Due to their complexity in terms of electronic structures, open-shell first-row transition metal complexes are difficult to be correctly described by most functionals. In this work, totally 19 reactions involving V, Cr, Mn, Fe, Co, Ni complexes, either monometallic or bimetallic, were used as testing set for 18 functionals ranging from generalized gradient approximation (GGA) to doubly-hybrid functionals, with experimental electron affinities and ligand association energies as standard. It is shown that for monometallic complexes PBE0-D3BJ and B3LYP-D3BJ perform the best, whereas MN15 and MN15L are the optimal functionals for bimetallic compounds. On the other hand, the accuracy of DLPNO-CCSD(T) is not significantly better than the best-performing functionals, and the use of doubly-hybrid functionals is risky.
Yumiao Ma
Computational Chemistry to the Heaven.